病毒广泛分布于自然界,对生物的健康和多样性产生深远的影响。由于传统分离方法的限制,大多数病毒仍然是未知的且难以培养。近年来,随着下一代测序(NGS)技术的迅猛发展,大量的宏基因组和宏转录组研究发现了海量的病毒基因序列,为深入探讨病毒的进化、多样性和流行病学提供了宝贵的信息。然而,由于样本中病毒的丰度较低,大部分鉴定到的病毒很难得到完整的病毒基因组序列。
近日,365英国上市公司在线彭友松团队在生物信息学国际权威期刊《Briefings in Bioinformatics》(IF2023=9.5)在线发表题为“VIGA: a one-stop tool for eukaryotic virus identification and genome assembly from next-generation-sequencing data”的研究论文,该研究开发了一种基于宏转录组和宏基因组数据对真核生物病毒基因组序列进行鉴定和组装的生物信息学工具VIGA,为鉴定和表征病毒组提供了帮助。

图1.发表封面图
VIGA由四个模块组成,包括病毒鉴定模块(Identification)、分类注释模块(Taxonomic annotation)、组装模块(Assembly)和新病毒发现模块(Novel virus discovery),可在Github上获取(https://github.com/viralInformatics/VIGA)。
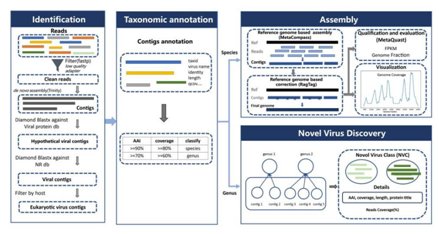
图2. VIGA的工作流程
该研究通过三种数据集:模拟病毒群落数据集、真实的宏基因组和宏转录组以及病毒株层次感染数据集,对VIGA的性能进行了深入评估,并将其与两种不同类型的工具进行了比较:基于参考基因组的工具(MetaCompass和VirGenA),以及从头组装的工具(Trinity和Haploflow)。
在模拟病毒群落数据集上,VIGA在恢复六种病毒的基因组完整性方面表现卓越,相较于其他方法具有更高的病毒基因组完整度。值得注意的是,VIGA的定量与复杂病毒群落中真实病毒丰度的相关性最高,进一步证实了其在病毒定量方面的有效性。
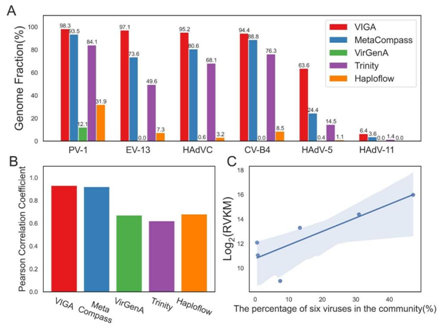
图3. VIGA及其他四种方法在模拟病毒群落数据集中的表现
VIGA在甘薯宏转录组数据集上的评估显示,VIGA组装出了10种中的9种病毒,基因组完整度分布在1.5%至100%之间,中位数为47.9%,高于其他软件工具。其中VIGA和MetaCompass成功组装了五种病毒的近乎完整基因组(完整度大于98%),其中三种也被Trinity组装出近完整基因组。在鸟粪宏基因组数据集中,总计有3种病毒的16个病毒株。在大多数样本中,VIGA成功组装了高比例的病毒基因组,中位数为86.54%,只有两个例外未成功组装基因组。
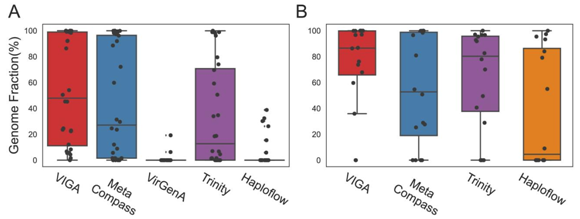
图4. VIGA及其他四种方法在模真实的宏转录组(左)和宏基因组(右)中的表现
在HIV数据集中(图5A),包括三种HIV毒株,它们之间的序列相似度为95%。VIGA在基因组完整度和菌株精准度指标上取得了近乎完美的表现,平均基因组完整度为98.20%,菌株精准度为100%。在组装错配方面,VIGA每100 kbp有2787个错配,略高于其他方法。在HBV数据集中,包含两个HBV毒株,它们之间的序列相似度为89%。VIGA在所有方法中表现最好,平均基因组完整度为99.91%,菌株精准度为100%,每100 kbp有1890.7个错配。
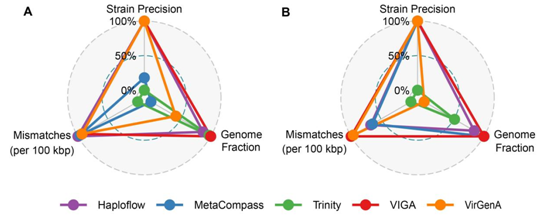
图5. VIGA及其他四种方法在病毒株水平上的表现
为了说明VIGA在大型数据集上的应用,我们使用VIGA重新分析人类微生物组项目(HMP)的1321个宏转录组样本。我们从467个样本中总共鉴定出125种已知的真核病毒,共组装了44种完整度较高的病毒(基因组完整度>80%),并发现疾病样本中的病毒组组成与健康人的病毒组组成不同。例如,克罗恩病的16种病毒中只有9种在健康人群中也被观察到。而对于每个疾病组,都有一种或多种疾病特异性病毒。例如,此前曾被认为与腹泻等肠道疾病的发生有关的轮状病毒A在克罗恩病患者中的丰度很高,中位数为4837 FPKM。
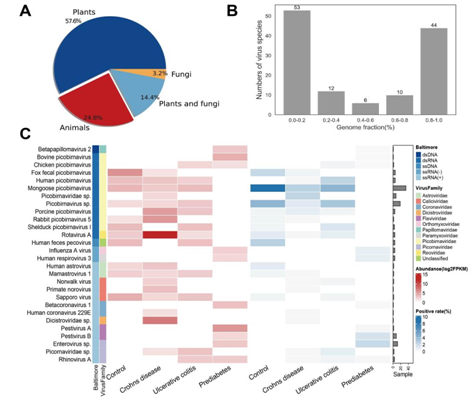
图6. HMP数据集中病毒组的鉴定和表征
该论文的第一作者为365英国上市公司在线的博士研究生傅萍,通讯作者为365英国上市公司在线生物信息中心、医学病毒学湖南省重点实验室的教授彭友松。这项工作得到了国家重点研发计划(2022YFC2303802)和国家自然科学基金(32170651 & 32370700)的支持。
论文链接:
https://doi.org/10.1093/bib/bbad444
论文信息:
Fu P, Wu Y, Zhang Z, Qiu Y, Wang Y, Peng Y. VIGA: a one-stop tool for eukaryotic virus identification and genome assembly from next-generation-sequencing data. Briefings in Bioinformatics. 2023 Nov 22;25(1):bbad444. doi: 10.1093/bib/bbad444. PMID: 38048079; PMCID: PMC10753531.